GV-971
GV-971 is a mixture of oligosaccharides with the degree of polymerization (dp) from 2 to 10. The non-clinical study showed that GV-971 penetrates the blood-brain-barrier (BBB) in its original form. Type 1 glucose transporter (GLUT1) was identified as one of the transporters accounting for its penetration mechanism of BBB. GV-971 could directly bind to multiple subregions of Aβ to inhibit Aβ fibril formation and destabilize the preformed fibrils into non-toxic monomers. Through targeting Aβ, GV-971 promoted microglia-mediated Aβ phagocytosis in vitro, and reversed cognition impairment in multiple AD models. All the preclinical pharmacology, PD/PK and toxicology studies were completed.
GV-971 exhibited a safe and well-tolerated profile in both phase 1 and phase 2 trials. In the recently completed phase 3 trial (NCT02293915), GV-971 was demonstrated to meet the primary endpoint, with statistic significance (p
Animals
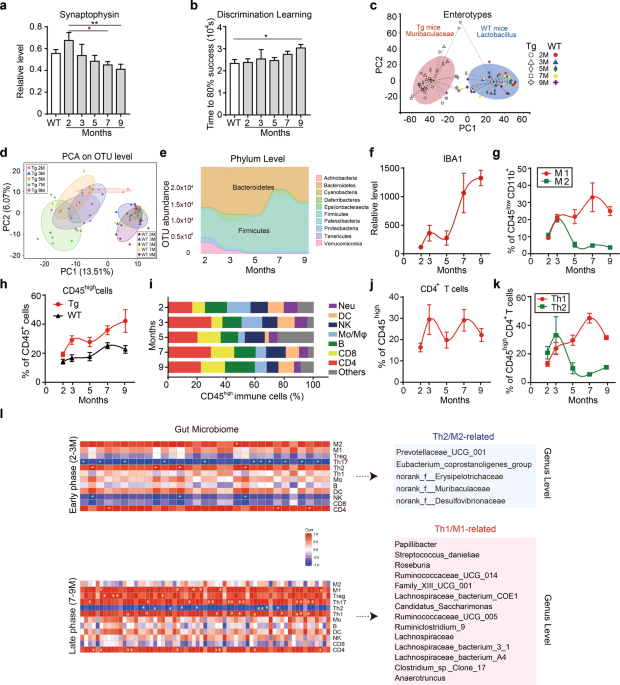
5XFAD (Tg) mice and co-housed WT mice (the corresponding WT mice generated by mating Tg mice and C57 mice) were bred in the same cage after birth. The WT mice (C57) were bred in separate cages to avoid microbiota cross transfer. All the mice were maintained in a room at 23 °C under a 12-hour (h) light-dark cycle. Mice were randomly allocated to different groups before treatment. For time course analysis of Tg mice, male and female Tg mice were sacrificed at 2-, 3-, 5-, 7- and 9-month old. Mouse brains were collected and stained for immune cell analysis and cytokine analysis. Before the mice were sacrificed, feces were collected for gut microbiota analysis. For GV-971 treatment, at 7-month old, Tg mice were treated with GV-971 at 100 mg/kg dose by oral gavage once daily for one month based on 450 mg twice per day in the Phase 3 trial. A behavioral test was performed to monitor cognitive activity after the last treatment. Then, the mouse brains and feces were used for different analyses. For behavior test, Tg mice and WT mice at different months as well as mice after treatment were tested by discrimination learning, as reported previously.57 For phenylalanine and isoleucine intraperitoneal injection treatment, 4-month-old C57 mice were treated with phenylalanine or isoleucine at 50 mg/kg for 4 days.
For Morris water maze (MWM) and Y maze tests, APP/PS1 mice were dosed orally with GV-971 at 50 and 100 mg/kg/day beginning at 9 months. Two tests were carried out at 13 months. For APP/PS1 mice time point analysis, feces were collected at various months of age before we sacrificed the mice for immune cell analysis. The mice and age-matched wild-type mice were sacrificed at 3-, 9-, and 14-month old for immune cell analysis. Mouse brains and blood were collected for different analyses. For APP/PS1 mice treated with GV-971 and antibiotics, 6-month-old mice were treated with GV-971 at 50 mg/kg by oral gavage once daily with or without antibiotic treatment for 2 months. The animal experiments were approved by the ethical committee of Shanghai SIPPR-Bk Lab Animal Co., Ltd (Number: 2016002) and by the Institutional Animal Care and Use Committee at Shanghai Institute of Materia Medica, China.
Behavioral tests
The MWM test is used to measure spatial learning and memory according to a protocol published previously.58 Briefly, the mice underwent 6-day acquisition experiments, and each mouse performed 3 trials each day. The animals were released into the water at the desired start position, and the latency to find the platform was timed. On the 7th day, the platform was removed, and the mice were allowed to swim for 60 s. The trace and the number of platform-site crossovers were recorded using a video camera.
Working memory was assessed by the Y maze according to the literature with some modifications.58 The Y maze was composed of three identical arms (A, B, C) with different cues. Mice were placed in the start arm (A) and the sequence of explored arms was recorded (such as ABCBA). The total number of arm entries and alternation behavior were recorded using a video camera. The accuracy of the Y maze was the ratio between the correct alternation and the total alternation.
Discrimination learning. 5XFAD mice and WT mice at different months as well as mice after treatment were tested by Cognition Wall discrimination spatial learning task, as reported previously.57 Mice were habituated to individual cages one week before the test. Approximately, 6 h before the test (10:00 am), the mice were housed in the automated PhenoTyper (HomeCage) to habituate to the cage. During the habituation phase, the mice had free access to water but limited access to food. The task began at 4:00 pm and continued during the night for 16 h. During the task, mice need to learn to enter the Cognition Wall through the left entrance (three entrances were offered) to obtain food (one food pellet was rewarded for every fifth entry through the left entrance). The total time needed to reach a criterion of 80% correct, computed as a moving window with window size equal to 30 (i.e., 24 correct entries out of the 30 last entries), was used as a measure during the task. All mouse movements were recorded by a computerized tracking system (Noldus, Ethovision). More time to reach the 80% correct criterion reflects less discriminative ability.
Faecal sample DNA extraction, PCR amplification and sequencing
All faecal samples were frozen at −80 °C before DNA extraction and analysis. The following steps were conducted by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). Microbial DNA was extracted from faecal samples using the E.Z.N.A.® Soil DNA Kit (Omega Bio-Tek, Norcross, GA, U.S.) according to the manufacturer’s protocols. The final DNA concentration and purification were determined by a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA), and DNA quality was checked by 1% agarose gel electrophoresis. The V3-V4 hypervariable regions of the bacterial 16S rRNA gene were amplified with primers 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806 R (5′-GGACTACHVGGGTWTCTAAT-3′) by a thermocycler PCR system (GeneAmp 9700, ABI, USA). PCR reactions were conducted using the following program: 3 min (min) of denaturation at 95 °C, 27 cycles of 30 sec (s) at 95 °C, 30 s for annealing at 55 °C, and 45 s for elongation at 72 °C, and a final extension at 72 °C for 10 min. PCR reactions were performed in triplicate in a 20 μL mixture containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mmol/L dNTPs, 0.8 μL of each primer (5 μmol/L), 0.4 μL of FastPfu Polymerase and 10 ng of template DNA. The resulting PCR products were extracted from a 2% agarose gel and further purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) and quantified using QuantiFluor™-ST (Promega, USA) according to the manufacturer’s protocol. Purified amplicons were pooled in equimolar and paired-end sequenced (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego, USA).
Processing of sequencing data
The following steps were conducted by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). Raw fastq files were demultiplexed, quality filtered by Trimmomatic and merged by FLASH based on the following criteria: (i) The reads were truncated at any site that received an average quality score http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The taxonomy of each 16S rRNA gene sequence was analyzed by the RDP Classifier algorithm (http://rdp.cme.msu.edu/) against the Silva 16S rRNA database (silva 132/16s bacteria) using a confidence threshold of 70%.
Metabolites sample preparation
Samples stored at −80 °C were taken out and thawed at room temperature. The following steps were conducted by Majorbio Bio-Pharm Technology Co., Ltd (Shanghai, China). In the experiment, 50 mg samples were used, and 400 μL methanol-water (4:1, v/v) were also added to homogenize the sample using a homogenizer for 10 s. The solution was ultrasonically extracted on ice for 10 min and stored at −20 °C for 30 min, then centrifuged for 15 min at 13000 rpm at 4 °C. For LC-MS analysis, 200 μL supernatant was used. QC sample was prepared by mixing aliquots of all samples to be a pooled sample and then analyzed using the same method with the analytic samples. The QCs were injected at regular intervals (every 10 samples) throughout the analytical run to provide a set of data from which repeatability can be assessed.
LC/MS analysis parameters
The following steps were conducted by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). LC-MS was performed on AB Sciex TripleTOF 5600TM mass spectrometer system (AB SCIEX, USA). LC Conditions: Column: Acquity BEH C18 column (100 mm × 2.1 mm i.d., 1.7 µm; Waters, Milford, USA). Solvent: The column was maintained at 40 °C and separation was achieved using the following gradient: 5% B–30% B over 0–3 min, 30% B–95% B over 3–9 min, 95% B–95% B over 9–13.0 min; 95% B–5% B over 13.0–13.1 min, and 13.1–16 min holding at 5 % B at a flow rate of 0.40 mL/min, where B is acetonitrile: isopropanol 1:1 (0.1% (v/v) formic acid) and A is aqueous formic acid (0.1% (v/v) formic acid). Injection Volume was 20 μL. The mass spectrometric data was collected using an AB Sciex TripleTOF 5600TM mass spectrometer system equipped with an electrospray ionization (ESI) source operating in either positive or negative ion mode with a capillary voltage 1.0 kV, sample cone, 40 V, collision energy 6 eV. The source temperature was set at 120 °C, with a desolvation gas flow of 45 L/h. Centroid data was collected from 50 to 1000 m/z with a 30000 resolution.
In vitro differentiation of naïve CD4+T to Th1 and Th2 induced by the supernatant of mice feces
We used a previously well-established protocol for the in vitro culture of fecal samples and collected the supernatant.44,45,46 Briefly, gut feces of 7-month-old mice were collected, put into the culture medium (with or without GV-971, final concentration of 100 µg/mL) and 96 deep-well plates (2 mL total volume) at 37 °C on the shaking bed (500 rpm) inside the anaerobic incubator for a total culture period of 24 h before we collected the gut-microbiota-derived metabolites-containing supernatant. Naïve CD4+ T cells were separated from the splenocytes of 8-month-old C57BL/6 female mice using the naïve CD4+ T cell Isolation Kit (Stem Cell, Cat No. 19765). A total of 0.5 × 106 cells/well in 0.5 mL of RPMI-1640 medium were plated in 48-well anti-CD3 and anti-CD28 pre-coated plates, and the culture medium was supplemented with cytokines and blocking antibodies. Th0: 20 ng/mL mIL-2; Th1: 20 ng/mL mIL-2, 10 μL supernatant, 5000 ng/mL 11B11; Th2: 20 ng/mL mIL-2, 10 μL supernatant, 5000 ng/mL XMG1.2. After incubation at 37 °C in 5% CO2 for 5 days, cells were stimulated with Cell Activation Cocktail (Thermo fisher, #00-4975-03) in 5% CO2 at 37 °C for 4 h, washed twice with PBS, labeled with zombie yellow (Biolegend, # 423104) to exclude dead cells. Non-specific binding of immunoglobulin to the Fc receptors was blocked with anti-CD16/32 (Biolegend, #101320). Intracellular staining was performed with anti-CD4 (Biolegend, #100406), anti-IFNγ (Biolegend, #505826) and anti-IL4 (Biolegend, #504104) according to the manufactures’ protocal. Cells were acquired on a BD Aria III cytometer, and data were analyzed using FlowJo software.
Immunohistochemistry
For 3,30-diaminobenzidine (DAB)-developed staining, sections were immunostained using a Leica BOND-RX Autostainer (Leica Microsystems) and Coverslipper CV5030 (Leica Microsystems) according to the manufacturer’s IHC protocol. Briefly, sections were submitted to heat-induced epitope retrieval with Epitope Retrieval solution 2 (ER2, AR9640, Leica Biosystems) for 20 min. Endogenous peroxidase activity was blocked with 3%-4% (v/v) hydrogen peroxide (part of DS9800, Leica Biosystems) for 10 min. Then, sections were incubated with blocking buffer (10% NGS in PBS with 0.3% Triton x-100) for 60 min. Finally, staining was performed using the Bond Polymer Refine Detection System (DS9800, Leica Biosystems) according to the manufacturer’s protocol. The primary antibody incubation time was 30 min. Sections were stained for activated microglia using rabbit anti-IBA1 antibody (1:1,000, cat# 019-19741, Wako), amyloid depositions using mouse anti-Aβ42 antibody (1:1,000; cat# 803003, Biolegend) and Tau phosphorylation using mouse anti-PHF-Tau & tangles -Thr231 antibody (1:300, cat# MN1040, Thermo Fisher). Stained slices were automatically scanned by a high-throughput bright field scanner (NanoZoomer 2.0HT, Hamamatsu), and images were obtained by NDP.scan 3.2 software (Hamamatsu). For fluorescent staining, slides were blocked by blocking buffer (10% NGS in PBS with 0.3% Triton x-100) at RT for 1 h, and then incubated in the primary antibody solution (Iba-1 1:1,000, Aβ42 1:1,000, Tau 1:300) overnight at 4 °C. After washing, slides were incubated with fluorescent anti-rabbit or anti-mouse secondary antibody (1:1000, Invitrogen) for 60 min at RT and further washed 3 times in PBS. Finally, slides were counterstained with DAPI (1:10000 in PBS, Sigma) for 5 min at RT, washed, sealed and stored at 4 °C for image acquisition. Representative fluorescence images were acquired by upright fluorescence microscope Zmager-m2 (Zeiss, Germany) under 10× objective using Zen software (Zeiss).
Amino acid detection
A set of amino acid standard mixture solutions was prepared at a concentration range of 100–2000 μmol/L. A portion of 10 μL of each standard mixture solution or plasma sample was pipetted into the bottom of a tube, and then 70 μL of sodium borate buffer (200 mmol/L at pH 8.8) was added. After 20 μL of 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) (4 mg/mL in acetonitrile) was added, the tube was closed and heated for 10 min at 55 °C to form AQC–amino acid. The solution was then cooled down to room temperature and 2 μL portion of each solution was injected into the UPLC-ESI-MS system for amino acid analysis without further purification.
Human subjects
The blood from MCI due to AD patients and healthy control was collected from a pilot study. MCI due to AD is defined in the NIA-AA 2011 clinical and research diagnostic criteria for MCI due to AD. The patients with MCI due to AD in this study must meet the following criteria. First, concern regarding a change in cognition. Second, impairment in one or more cognitive domains. Third, preservation of independence in functional abilities. Forth, not demented (NIA-AA 2011 clinical diagnosis criteria for MCI due to AD). All participants underwent a screening process that included a review of their medical history, physical and neurological examinations, laboratory tests, and MRI scans. The clinical assessment of mild cognitive impairment or dementia included neuropsychological tests, as well as behavioral and psychiatric interviews conducted by attending psychiatrists. AD patients recorded scores of
The sample size for the first cohort (Fig. 5h, i) is 9 MCI due to AD patients and 18 healthy subjects. The sample size for the second cohort (Fig. 5j) is 22 MCI due to AD patients and 22 healthy subjects. A diagnosis of MCI was based on the following criteria, which were adapted from the MCI diagnostic criteria of Petersen: (1) memory complaints, preferably corroborated by a spouse or relative; (2) objective memory impairment; (3) normal general cognitive function; (4) intact activities of daily living; and (5) absence of dementia. The Ethics Committee of the Shanghai Mental Health Centre Institutional Review Board approved the study (Number: 2016-22R1). Informed consents were obtained from the subjects and the guardian of the subjects. Information about gender and age etc. are provided below.
First cohort
|
Type |
Sample ID |
Age |
Gender |
|---|---|---|---|
|
Healthy |
HC-01 |
75 |
F |
|
Healthy |
HC-02 |
71 |
F |
|
Healthy |
HC-04 |
69 |
F |
|
Healthy |
HC-05 |
73 |
F |
|
Healthy |
HC-08 |
63 |
M |
|
Healthy |
HC-09 |
62 |
M |
|
Healthy |
HC-10 |
68 |
M |
|
Healthy |
HC-11 |
61 |
M |
|
Healthy |
HC-12 |
68 |
M |
|
Healthy |
HC-13 |
75 |
F |
|
Healthy |
HC-14 |
70 |
F |
|
Healthy |
HC-15 |
68 |
M |
|
Healthy |
HC-16 |
62 |
M |
|
Healthy |
HC-17 |
75 |
M |
|
Healthy |
HC-18 |
65 |
F |
|
Healthy |
HC-19 |
64 |
F |
|
Healthy |
HC-20 |
64 |
F |
|
Healthy |
HC-22 |
63 |
F |
|
MCI due to AD |
MCI-02 |
61 |
F |
|
MCI due to AD |
MCI-03 |
79 |
M |
|
MCI due to AD |
MCI-04 |
71 |
F |
|
MCI due to AD |
MCI-05 |
72 |
F |
|
MCI due to AD |
MCI-06 |
80 |
F |
|
MCI due to AD |
MCI-07 |
65 |
M |
|
MCI due to AD |
MCI-09 |
61 |
M |
|
MCI due to AD |
MCI-10 |
69 |
M |
|
MCI due to AD |
MCI-14 |
77 |
M |
Second cohort
|
Category |
Sample ID |
Age |
Gender |
|---|---|---|---|
|
Healthy |
HC-0149 |
73 |
M |
|
Healthy |
HC-0150 |
70 |
F |
|
Healthy |
HC-0153 |
63 |
F |
|
Healthy |
HC-0156 |
67 |
M |
|
Healthy |
HC-0158 |
71 |
F |
|
Healthy |
HC-0165 |
61 |
F |
|
Healthy |
HC-0168 |
62 |
F |
|
Healthy |
HC-0172 |
70 |
F |
|
Healthy |
HC-0176 |
71 |
F |
|
Healthy |
HC-0187 |
73 |
F |
|
Healthy |
HC-0191 |
65 |
M |
|
Healthy |
HC-0013 |
70 |
F |
|
Healthy |
HC-0043 |
81 |
M |
|
Healthy |
HC-0050 |
64 |
F |
|
Healthy |
HC-0051 |
66 |
M |
|
Healthy |
HC-0056 |
75 |
M |
|
Healthy |
HC-0068 |
67 |
F |
|
Healthy |
HC-0074 |
57 |
F |
|
Healthy |
HC-0076 |
60 |
F |
|
Healthy |
HC-0081 |
71 |
F |
|
Healthy |
HC-0089 |
57 |
F |
|
Healthy |
HC-0090 |
76 |
F |
|
MCI due to AD |
MCI-0169 |
65 |
F |
|
MCI due to AD |
MCI-0175 |
61 |
F |
|
MCI due to AD |
MCI-0211 |
69 |
M |
|
MCI due to AD |
MCI-0229 |
65 |
M |
|
MCI due to AD |
MCI-0240 |
59 |
F |
|
MCI due to AD |
MCI-0244 |
64 |
F |
|
MCI due to AD |
MCI-0257 |
61 |
F |
|
MCI due to AD |
MCI-0260 |
60 |
F |
|
MCI due to AD |
MCI-0282 |
66 |
F |
|
MCI due to AD |
MCI-0284 |
64 |
F |
|
MCI due to AD |
MCI-0287 |
75 |
F |
|
MCI due to AD |
MCI-0303 |
62 |
F |
|
MCI due to AD |
MCI-0319 |
63 |
F |
|
MCI due to AD |
MCI-0332 |
67 |
F |
|
MCI due to AD |
MCI-0374 |
61 |
M |
|
MCI due to AD |
MCI-0385 |
68 |
F |
|
MCI due to AD |
MCI-0401 |
57 |
F |
|
MCI due to AD |
MCI-0414 |
64 |
F |
|
MCI due to AD |
MCI-0001 |
75 |
M |
|
MCI due to AD |
MCI-0043 |
55 |
F |
|
MCI due to AD |
MCI-0061 |
63 |
F |
|
MCI due to AD |
MCI-0091 |
78 |
F |
Th1 cell staining in human samples
Two milliliters of peripheral blood from healthy people or patients were treated with red blood cells lysis buffer. A total of 1 × 106 cells were collected and incubated in 100 μL of PBS containing Zombie Yellow Dye (Biolegend, #423104) for 15 min to label live cells. After washing, samples were stained with APC/Cy7-conjugated anti-human CD45 (Biolegend, #304014), Alexa Fluor 700-conjugated anti-human CD3 (Biolegend, #344822), FITC-conjugated anti-human CD4 (Biolgend, #300506), Brilliant Violet 421-conjugated anti-human CXCR3 (Biolgend, #353716) and APC-conjugated anti-human CCR6 (Biolgend, #353416) for 30 min. Stained cells were washed twice with PBS. Cells were acquired using FACS Fortessa X-20 flow cytometer (BD Biosciences, San Jose, CA). Live CD45+CD3+CD4+CXCR3+CCR6– cells were gated as Th1 subsets as previously described.59 Data analysis were performed with flowjo software (version 10.5, LLC)
In vitro differentiation induced by phenylalanine and isoleucine
As previously described,60 naïve CD4+ T cells were enriched from the splenocytes of 8-month-old C57BL/6 female mice using the Naïve CD4+ T cell Isolation Kit (Stem Cell, #19765). For cell differentiation assay, a total of 1 × 105 cells/well in 0.2 mL of RPMI-1640 medium were plated in anti-CD3/CD28-coated 96-well plates (anti-CD3, 2 μg/mL; anti-CD28, 1 μg/mL), and the culture medium was supplemented with cytokines or blocking antibodies. Th0: mIL-2 (10 ng/mL); Th1: mIL-2 (10 ng/mL) and 11B11 (5000 ng/mL). Phenylalanine (final concentration, 1 mmol/L), isoleucine (final concentration, 1 mmol/L) and GV-971 (final concentration, 100 µg/mL) were added into the indicated wells, respectively. After incubation at 37 °C in 5% CO2 for 5 days, cells were stimulated with Cell Activation Cocktail (Thermo fisher, #00-4975-03) in 5% CO2 at 37 °C for 4 h, washed twice with PBS, and labeled with zombie yellow (Biolegend, #423104) to exclude dead cells. Non-specific binding of immunoglobulin to the Fc receptors was blocked with anti-CD16/32 (Biolegend, #101320). Intracellular staining was performed with anti-CD4 (Biolegend, #100406) and anti-IFNγ (Biolegend, #505830) according to the manufactures’ protocal. Cells were acquired on a BD Fortessa cytometer, and data were analyzed using FlowJo software.
In vitro proliferation induced by phenylalanine and isoleucine
A total of 1 × 106 naïve CD4+ T cells were dissolved with 3 mL of Th1 differentiation Medium (10 ng/mL of mIL-2, 20 ng/mL of mIL-12 and 5000 ng/mL of 11B11) and plated in anti-CD3/CD28-coated 60-mm culture dish (anti-CD3, 2 μg/mL; anti-CD28, 1 μg/mL). The cells were incubated in 5% CO2 at 37 °C for 5 days.
Next, flow cytometry was applied to gate live cells (Zombie yellow dye, Biolegend, #423104) and CD4+CXCR3+ Th1 cells were sorted by anti-CD4 (Biolegend, #100406) and anti-CXCR3 (Biolegend, #126522) staining. The viability of sorted Th1 cells was further confirmed by Trypan blue staining.
The sorted Th1 cells was stained with CellTrace (Thermo Fisher, #C34557) and plated in anti-CD3/CD28-coated microplate and cultured in RPMI-1640 medium in the presence of phenylalanine (final concentration, 1 mmol/L), isoleucine (final concentration, 1 mmol/L) and GV-971 (final concentration, 100 µg/mL). After incubation in 5% CO2 at 37 °C for 4 days, cells were stimulated with Cell Activation Cocktail (Thermo fisher, #00-4975-03) in 5% CO2 at 37 °C for 4 h, washed twice with PBS, and labeled with zombie yellow (Biolegend, #423104) to exclude dead cells. Non-specific binding of immunoglobulin to the Fc receptors was blocked with anti-CD16/32 (Biolegend, #101320). Intracellular staining was performed with anti-CD4 (Biolegend, #100422) and anti-IFNγ (Biolegend, #505806) according to the manufactures’ protocal. Cells were acquired on a BD Fortessa cytometer, and data were analyzed using FlowJo software.
Uptake of phenylalanine
Naïve CD4+ T cells were separated from the splenocytes of 8-month-old C57BL/6 female mice using the Naïve CD4+ T cell Isolation Kit (Stem Cell, Cat No. 19765) and were induced to Th1 differentiation by 20 ng/mL IL-12. After 3 days, a total of 5 × 105 cells/well Th1 cells in 0.5 mL of RPMI-1640 medium were plated into 48-well plates. 13C-labelled phenylalanine and 5 mM amino transporter inhibitor BCH (CAS:20448-79-7) were added into indicated wells. After 0.5 h, cells were collected and washed twice with ice-cold D-PBS. Then, 50 µL deionized water was added and cells were lysed through freezing and thawing twice at −80 °C. The cell lysate was centrifuged at 12000 × g for 10 min and 13C-labelled phenylalanine in the supernatant was analyzed by Mass spectrometry.
Antibiotic treatments
Mice were treated by adding an antibiotic solution (ABX) containing ampicillin (0.1 mg/mL, final concentration in drinking water), streptomycin (0.5 mg/mL, final concentration in drinking water), and colistin (0.1 mg/mL, final concentration in drinking water) (Sigma-Aldrich) to sterile drinking water. Solutions and bottles were changed 3 times and once weekly, respectively. The antibiotic activity was confirmed by 16S rRNA gene sequencing. Types of bacteria with a relative abundance of less than 0.01 are deleted in the Tg group. The duration of ABX treatment was slightly different based on the experimental settings. In the context of faecal microbia transplantation experiments, mice received 3 days of ABX before undergoing faecal microbia transplantation the next day by oral gavage using animal feeding needles.
FMT experiments
FMT was performed by thawing faecal material. Then, 200 μL of the suspension was transferred by oral gavage into each ABX-pretreated recipient. Twelve-month-old C57 mice were first treated with an antibiotic cocktail in drinking water for 3 days, and then 40 mg of the mixed stool suspended in PBS was inserted by gavage into each mouse 3 times with a 2-day break in between. After 3 days, 4.2 µg Aβ 1–40 oligomer was injected into both sides of the hippocampus. The mice were sacrificed 3 days later and used for different analyses.
Bioinformatics analysis
Principal component analysis (PCA) is a method that simplifies the complexity in high-dimensional data while retaining trends and patterns. PCA reduces data by geometrically projecting them onto lower dimensions called principal components (PCs), with the goal of finding the best summary of the data using a limited number of PCs.28 Principle coordinate analysis (PCoA) is the modified version of PCA analysis, in which different distance metrics (e.g., Bray-Curtis distances) can be used, compared to the Euclidean distances that is solely used in PCA. Both PCA and PCoA analysis used in this study were performed at Majorbio I-Sanger Cloud platform (www.i-sanger.com). For correlational analysis, trend changes in the abundance of bacteria were correlated with changes in the frequency of immune cells. The exact correlation coefficient and the p-value (set to 0.05) were calculated using the Pearson parametric correlation test using the R package “ggcorrplot”. Correlational circus maps were generated using the R package “igraph”. Pathway analysis and biological function enrichment analysis were performed using the Kyoto Encyclopedia of Gene Genotype (KEGG). Data were enriched using the R package “DOSE”, “GO.db”, “GSEABase” and “ClusterProfiler”. Only pathways with a false discovery rate (FDR) corrected p-value of (Wu M, Gu L (2019). TCseq: Time course sequencing data analysis. R package version 1.8.0). A standardized z-score transformation was applied to convert the fraction values to z-scores before analysis in all heatmaps. Heatmaps and volcano plots are also plotted using the R packages. The ROC biomarker analysis and random forest classification were performed with MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/). The Mouseac database used in Supplementary information, Fig. S2 is http://www.mouseac.org/. Other bioinformatics analyses, including the barplot representation of gut microbiota abundance at the family level, lists of significant changing bacteria, etc. were conducted using the online platform of the Majorbio I-Sanger Cloud (www.i-sanger.com).
Flow cytometry
Mice were anesthetized, blood samples were collected into EDTA-containing tubes, and red blood cells were removed using 1 × red blood lysis buffer. Before tissue collection, the brains were perfused with ice-cold PBS to avoid sampling the circulating blood immune cells, and the brains were removed, chopped into pieces and dissected according to the introduction of the Adult Brain Dissociation Kit (Miltenyi, Cat No. 130-107-677) using the gentleMACS dissociator (Miltenyi Biotec). The brain homogenate was filtered through a 70-μm cell strainer and centrifuged at 300 × g for 5 min at 4 °C. The cell pellet was resuspended in cold PBS buffer and centrifuged again at 300 × g for 5 min at 4 °C. All samples were counted and adjusted to a density of 2–3 × 106/100 μL, labeled with a Live/Dead kit for 30 min, and then centrifuged at 500 × g for 3 min at 4 °C. The cells were resuspended in 100 μL PBS buffer, blocked with anti-CD16/32 (Biolegend, Cat No. 101320) for 10 min, and incubated with the antibody according to the manufacturers’ protocols at 4 °C for 30 min. The following antibodies were used in the FACS analysis: CD45(30-F11)-APC-Cy7(103116, Biolegend), CD11b(M1/70)-FITC (101205, Biolegend), CX3CR1(SA011F11)-PE-Dazzle 594 (149014, Biolegend), Siglec-H-BV421 (566581, BD), F4/80(BM8)-BV421 (123132, Biolegend), CD86(IT2.2)-PE (305438, Biolegend), CD206(15-2)-APC (321110, Biolegend), CD206(15-2)-BV785 (321142, Biolegend), CD11c(N418)-PE-Cy7 (117318, Biolegend), CD8(53-6.7)-Percp-Cy5.5 (100734, Biolegend), Ly-6C(HK1.4)-PE-Dazzle 594 (128044, Biolegend), Gr-1(RB6-8C5)-Percp-Cy5.5 (108428, Biolegend), B220(RA3-6B2)-BV421 (103240, Biolegend), CD19(6D5)-PE (115508, Biolegend), CD49b(DX5)-PE-Cy7 (108922, Biolegend), CD4(GK1.5)-PE-Cy7 (100422, Biolegend), CD4(GK1.5)-FITC (100406, Biolegend), CXCR3(CXCR3-173)-BV421 (126522, Biolegend), CCR4(L291H4)-PE-Cy7 (359410, Biolegend), CCR6(29-2L17)-APC (129814, Biolegend), CD127(A019D5)-PE (351304, Biolegend), CD25(3C7)-Percp-Cy5.5 (101912, Biolegend), Live/Dead (423104, Biolegend). Cells were added to 500 μL PBS buffer, centrifuged at 500 × g for 3 min at 4 °C and resuspended in 200 μL running buffer. Relevant negative control, Fluorescence Minus One (FMO) control and each fluorescence compensation sample were used to adjust fluorescence compensation and identify the populations of interest. Cells were acquired on a BD Aria III cytometer, and data were analyzed using FlowJo software.
Antibody array
The following steps were conducted by RayBiotech (Guangzhou, China). The brain homogenates (from 20 mg tissue) were analyzed with a glass slide-based antibody cytokine array including 200 proteins (RayBiotech, GSM-CAA-4000-1). A 100 μL sample diluent was added to each well and incubated at room temperature for 30 min. Then, the buffer was replaced with another 100 μL of sample and incubated at room temperature for 2 h. The samples were discarded and washed 5 times (5 min each) with 150 μL of 1 × Wash Buffer I and 2 times (5 min each) with 150 μL of 1 × Wash Buffer II at room temperature with gentle shaking. After that, 80 μL of the detection antibody cocktail were added to each well and incubated at room temperature for 2 h. The slide was washed 5 times (5 min each) with 150 μL of 1 × Wash Buffer I and then 2 times with 150 μL of 1 × Wash Buffer II at room temperature with gentle shaking. Eighty microliters of Cy3 equivalent dye-conjugated streptavidin was added to each well and incubated at room temperature for 1 h. After 5 washes (5 min each), the signal was visualized through a laser scanner. The data were then visualized by a heatmap diagram (www.metaboanalyst.ca).
Brain section preparation
Mice were transcardially perfused with 0.9% NaCl after deep anesthesia with pentobarbital (100 mg/kg, i.p.). Brain tissues were quickly removed, frozen and stored at −70 °C. Serial coronal brain sections (12 μm thickness) were created using a sliding, freezing microtome (Leica Microsystems), mounted on slides and dried overnight in the air at room temperature. Tissue sections were stored at −80 °C or used immediately.
Laser microdissection and Q-PCR analysis
Frozen mouse brain samples were sectioned and collected on PEN membrane slides (Leica, 11600288). The hippocampus was isolated by laser microdissection microscopy (Leica Microsystems, LMD6). RNA was extracted with a RNeasy Micro Kit (Qiagen, 74004) and reverse transcribed into cDNA (Takara, PrimeScript RT Master Mix, RR036A). Q-PCR was performed using the ABI 7500 system via the SYBR method (Takara, SYBR Premix Ex Taq II). Following primers were used: Synaptophysin-forward: CAGTTCCGGGTGGTCAAGG; Synaptophysin-reverse: ACTCTCCG TCTTGTTGGCAC; actin-forward: GCTCTTTTCCAGCCTTCCTT; actin-reverse: AGGTCTTTACGGAT GTCAACG.
Statistical analysis
In the behavior test, animals were randomly distributed into different groups. For two group comparisons, an unpaired two-tailed Student’s t-test was applied. For significantly changing bacteria lists, we used the online platform of the Majorbio I-Sanger Cloud to perform Wilcoxon rank-sum test, and the P-value was based on a two-tailed test with FDR corrected, the significant level was set to 0.05, and the 0.95 confidence intervals were calculated through the bootstrap algorithm. For more than two group comparisons, one-way ANOVA or two-way ANOVA followed by Dunnett’s test was performed. All data with error bar are represented as mean ± SEM. P